Log2CPM Boxplots of Most Significant DE Genes
Last updated: 2025-02-09
Checks: 7 0
Knit directory: CX5461_Project/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20250129) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version dc74b1f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: data/Boxplot_CX_0.5_24.csv
Untracked: data/Boxplot_CX_0.5_3.csv
Untracked: data/Boxplot_CX_0.5_48.csv
Untracked: data/Boxplot_DOX_0.1_24.csv
Untracked: data/Boxplot_DOX_0.1_3.csv
Untracked: data/Boxplot_DOX_0.1_48.csv
Untracked: data/Boxplot_DOX_0.5_24.csv
Untracked: data/Boxplot_DOX_0.5_3.csv
Untracked: data/Boxplot_DOX_0.5_48.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/Log2CPM_Boxplots.Rmd) and
HTML (docs/Log2CPM_Boxplots.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | dc74b1f | sayanpaul01 | 2025-02-09 | wflow_publish("analysis/Log2CPM_Boxplots.Rmd") |
| html | cebe51b | sayanpaul01 | 2025-02-07 | Build site. |
| html | edd909b | sayanpaul01 | 2025-02-07 | Build site. |
| Rmd | 8230f77 | sayanpaul01 | 2025-02-07 | wflow_publish("analysis/Log2CPM_Boxplots.Rmd") |
📌 Log2CPM Boxplots of Most Significant DE Genes
This analysis generates boxplots for the log2CPM values of the most significant DE genes across CX-5461 and DOX treatments.
📌 Load Required Libraries
library(dplyr)Warning: package 'dplyr' was built under R version 4.3.2library(ggplot2)Warning: package 'ggplot2' was built under R version 4.3.3library(ggsignif)Warning: package 'ggsignif' was built under R version 4.3.1library(tidyr)Warning: package 'tidyr' was built under R version 4.3.3📌 CX_0.1_3 vs VEH_0.1_3
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_CX_0.1_3.csv"
output_boxplot <- "data/Boxplot_CX_0.1_3.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|CX.5461_0.1_3|VEH_0.1_3", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("CX.5461" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 100996485 2.032914 2.577945 10.55571 1.06e-17 1.52e-13 4.406806📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("CX.5461", Sample), "CX.5461", "VEH"),
Conc = "0.1",
Timepoint = "3"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
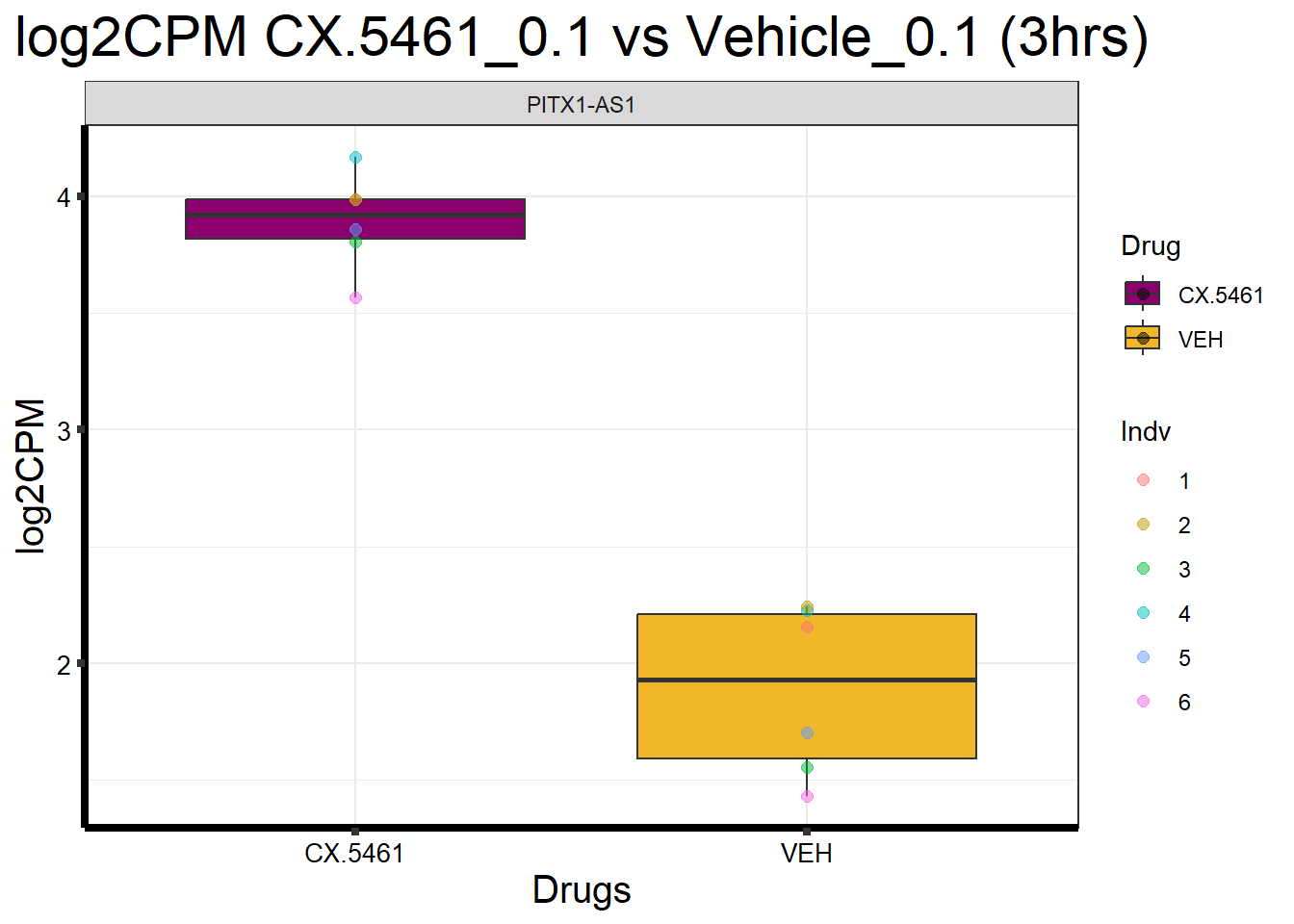
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 100996485 PITX1-AS1 17.3_CX.5461_0.1_3 4 CX.5461 0.1 3 4.166038
2 100996485 PITX1-AS1 84.1_CX.5461_0.1_3 5 CX.5461 0.1 3 3.859530
3 100996485 PITX1-AS1 84.1_VEH_0.1_3 5 VEH 0.1 3 1.704291
4 100996485 PITX1-AS1 90.1_CX.5461_0.1_3 6 CX.5461 0.1 3 3.566918
5 100996485 PITX1-AS1 90.1_VEH_0.1_3 6 VEH 0.1 3 1.431025
6 100996485 PITX1-AS1 17.3_VEH_0.1_3 4 VEH 0.1 3 2.229019📌Generate Boxplot
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM CX.5461_0.1 vs Vehicle_0.1 (3hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
| Version | Author | Date |
|---|---|---|
| edd909b | sayanpaul01 | 2025-02-07 |
📌 CX_0.1_24 vs VEH_0.1_24
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_CX_0.1_24.csv"
output_boxplot <- "data/Boxplot_CX_0.1_24.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|CX.5461_0.1_24|VEH_0.1_24", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("CX.5461" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 5347 -2.133241 3.460559 -10.514869 1.30e-17 1.86e-13 29.37176
2 10112 -2.657049 3.471443 -10.205466 5.94e-17 4.24e-13 27.86551
3 1062 -1.973517 2.975691 -9.795318 4.47e-16 2.11e-12 25.91921
4 100996485 1.869814 2.577945 9.738606 5.91e-16 2.11e-12 25.55081
5 9787 -2.084540 3.045935 -9.363571 3.75e-15 1.07e-11 23.88908
6 55635 -1.887472 3.616797 -9.263926 6.12e-15 1.37e-11 23.50584📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("CX.5461", Sample), "CX.5461", "VEH"),
Conc = "0.1",
Timepoint = "24"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
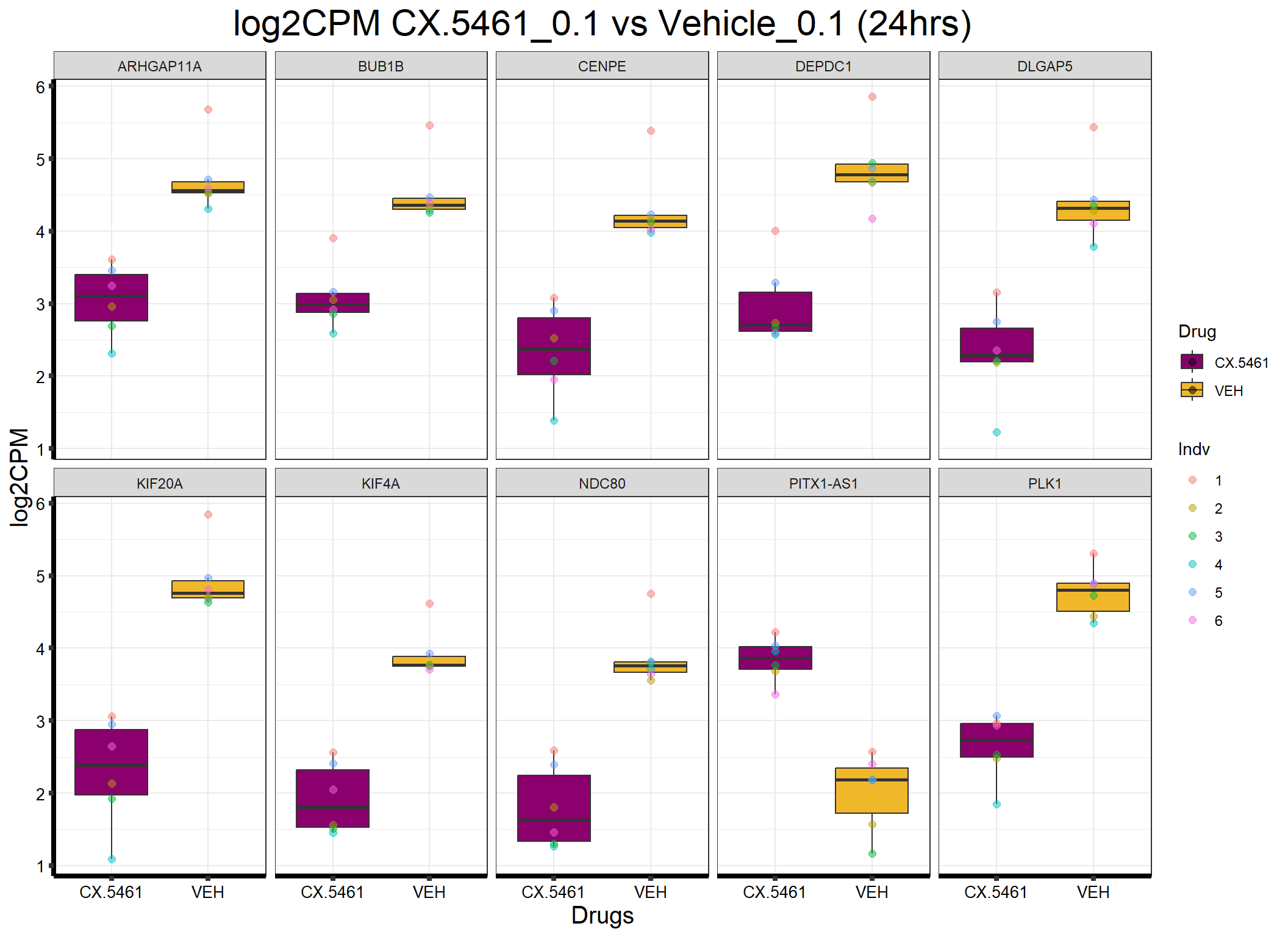
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 55635 DEPDC1 87.1_VEH_0.1_24 3 VEH 0.1 24 4.942031
2 55635 DEPDC1 17.3_VEH_0.1_24 4 VEH 0.1 24 4.691377
3 55635 DEPDC1 84.1_CX.5461_0.1_24 5 CX.5461 0.1 24 3.294382
4 55635 DEPDC1 84.1_VEH_0.1_24 5 VEH 0.1 24 4.869754
5 55635 DEPDC1 90.1_CX.5461_0.1_24 6 CX.5461 0.1 24 2.598657
6 55635 DEPDC1 90.1_VEH_0.1_24 6 VEH 0.1 24 4.176633📌Generate Boxplot
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM CX.5461_0.1 vs Vehicle_0.1 (24hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
| Version | Author | Date |
|---|---|---|
| edd909b | sayanpaul01 | 2025-02-07 |
📌 CX_0.1_48 vs VEH_0.1_48
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_CX_0.1_48.csv"
output_boxplot <- "data/Boxplot_CX_0.1_48.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|CX.5461_0.1_48|VEH_0.1_48", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("CX.5461" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 9787 -3.749715 3.045935 -14.24839 2.49e-25 1.50e-21 46.78947
2 5347 -3.292374 3.460559 -14.29479 2.02e-25 1.50e-21 47.13645
3 1062 -3.116017 2.975691 -14.19627 3.16e-25 1.50e-21 46.67902
4 55635 -3.247028 3.616797 -14.11906 4.50e-25 1.61e-21 46.41416
5 10112 -4.106672 3.471443 -13.42736 1.10e-23 3.15e-20 43.16052
6 9824 -2.534304 3.672008 -12.97567 9.16e-23 2.18e-19 41.25660📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("CX.5461", Sample), "CX.5461", "VEH"),
Conc = "0.1",
Timepoint = "48"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
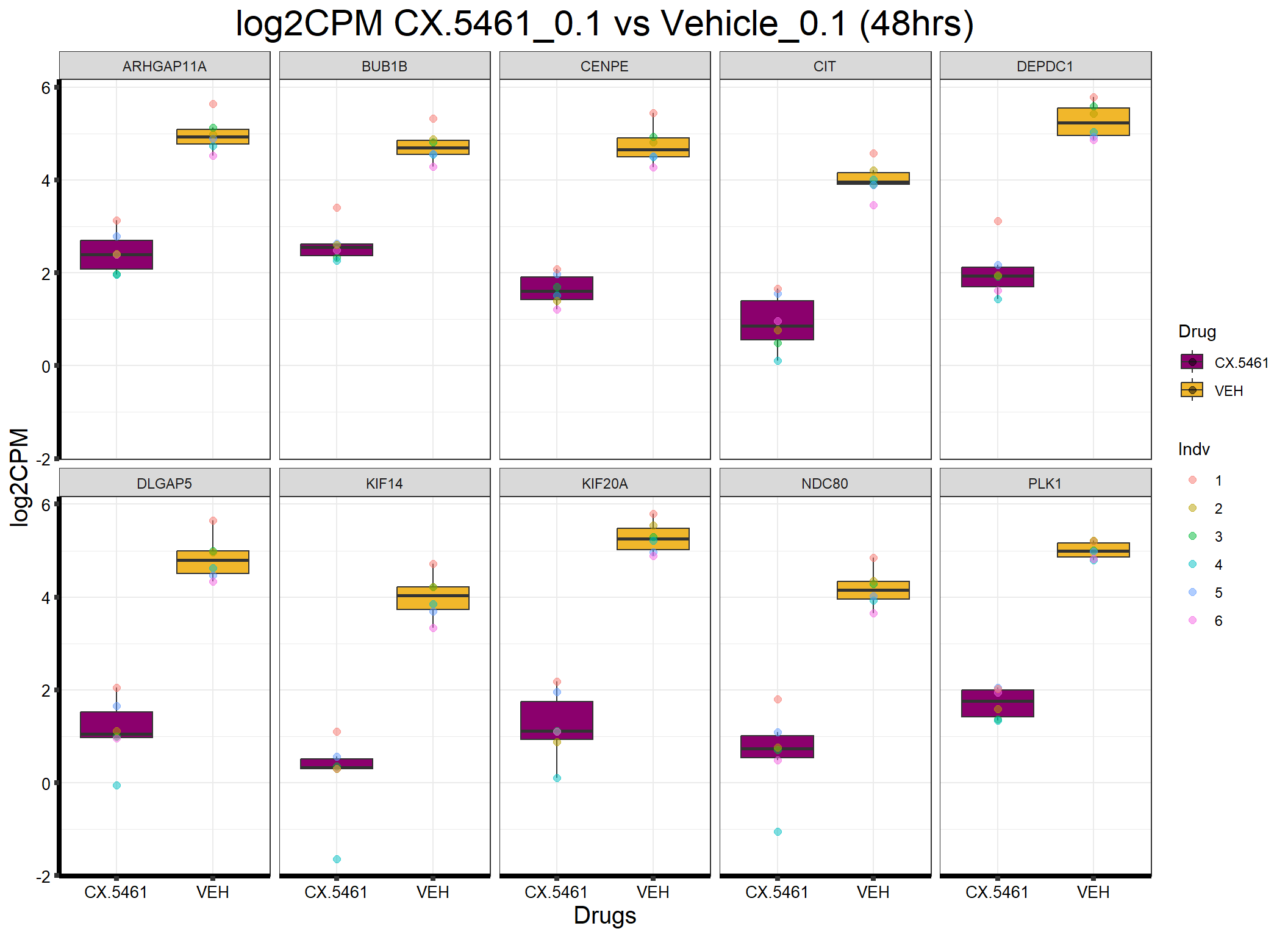
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 55635 DEPDC1 87.1_CX.5461_0.1_48 3 CX.5461 0.1 48 1.923754
2 55635 DEPDC1 87.1_VEH_0.1_48 3 VEH 0.1 48 5.592110
3 55635 DEPDC1 17.3_CX.5461_0.1_48 4 CX.5461 0.1 48 1.432382
4 55635 DEPDC1 17.3_VEH_0.1_48 4 VEH 0.1 48 5.042248
5 55635 DEPDC1 84.1_CX.5461_0.1_48 5 CX.5461 0.1 48 2.177464
6 55635 DEPDC1 84.1_VEH_0.1_48 5 VEH 0.1 48 4.930465📌Generate Boxplot
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM CX.5461_0.1 vs Vehicle_0.1 (48hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
| Version | Author | Date |
|---|---|---|
| edd909b | sayanpaul01 | 2025-02-07 |
📌 CX_0.5_3 vs VEH_0.5_3
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_CX_0.5_3.csv"
output_boxplot <- "data/Boxplot_CX_0.5_3.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|CX.5461_0.5_3|VEH_0.5_3", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("CX.5461" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 100996485 1.6268499 2.577945 8.383901 4.61e-13 6.580000e-09 1.830604
2 1106 -0.6992481 6.314996 -5.418152 4.53e-07 3.234724e-03 1.534904📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("CX.5461", Sample), "CX.5461", "VEH"),
Conc = "0.5",
Timepoint = "3"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
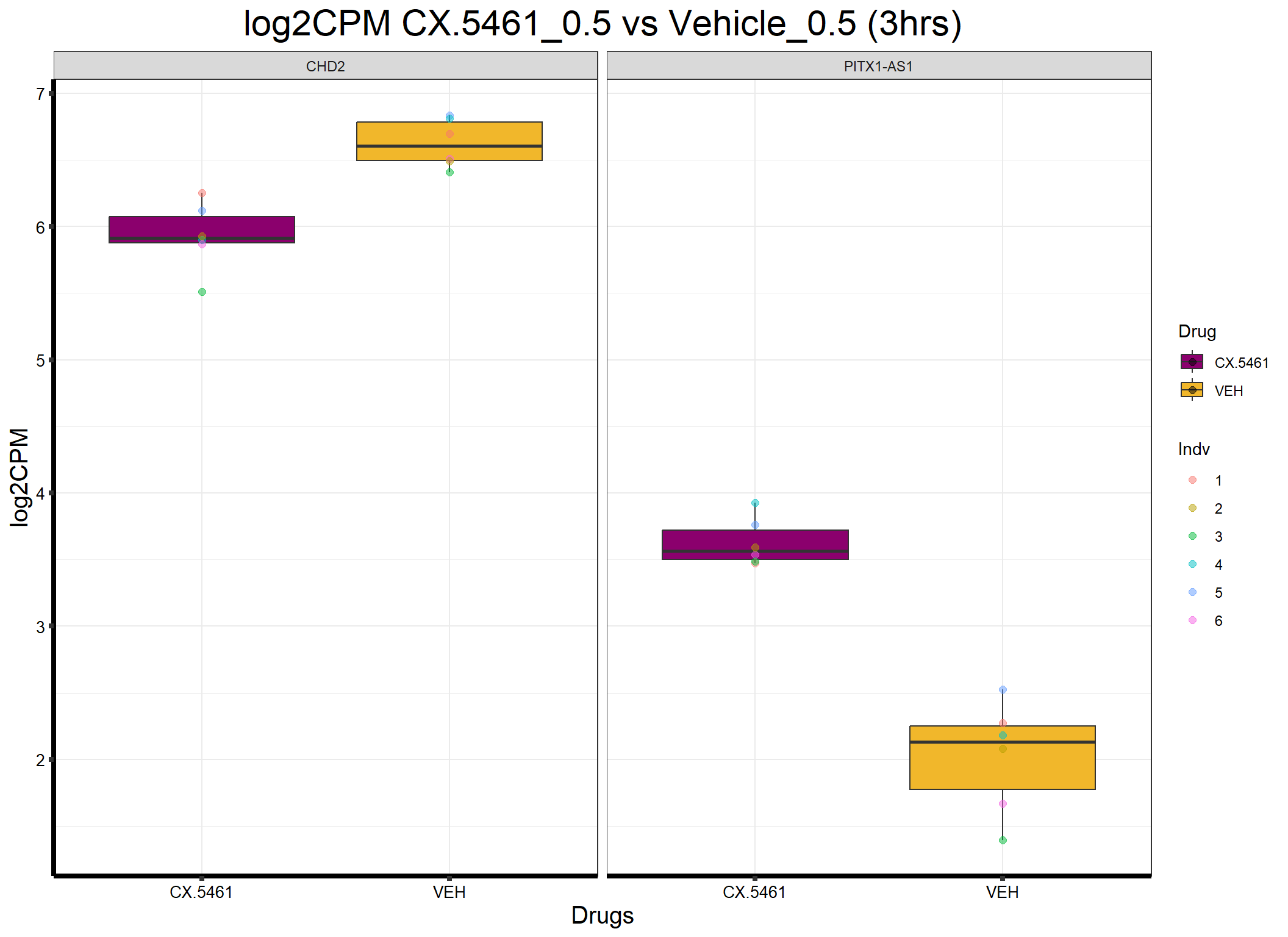
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 1106 CHD2 17.3_CX.5461_0.5_3 4 CX.5461 0.5 3 5.899468
2 1106 CHD2 84.1_CX.5461_0.5_3 5 CX.5461 0.5 3 6.120303
3 1106 CHD2 84.1_VEH_0.5_3 5 VEH 0.5 3 6.831808
4 1106 CHD2 90.1_CX.5461_0.5_3 6 CX.5461 0.5 3 5.868335
5 1106 CHD2 90.1_VEH_0.5_3 6 VEH 0.5 3 6.509929
6 1106 CHD2 75.1_CX.5461_0.5_3 1 CX.5461 0.5 3 6.254171📌Generate Boxplot
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM CX.5461_0.5 vs Vehicle_0.5 (3hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 CX_0.5_24 vs VEH_0.5_24
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_CX_0.5_24.csv"
output_boxplot <- "data/Boxplot_CX_0.5_24.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|CX.5461_0.5_24|VEH_0.5_24", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("CX.5461" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 1062 -2.585719 2.975691 -11.341720 2.29e-19 3.27e-15 33.27965
2 5347 -2.531512 3.460559 -11.132492 6.34e-19 4.53e-15 32.38951
3 10112 -3.015316 3.471443 -10.399545 2.29e-17 1.09e-13 28.90014
4 701 -1.691639 3.456542 -9.913932 2.49e-16 8.90e-13 26.67698
5 55635 -2.072008 3.616797 -9.464552 2.28e-15 4.07e-12 24.52676
6 9787 -2.287759 3.045935 -9.470036 2.22e-15 4.07e-12 24.49988📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("CX.5461", Sample), "CX.5461", "VEH"),
Conc = "0.5",
Timepoint = "24"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 55635 DEPDC1 87.1_VEH_0.5_24 3 VEH 0.5 24 4.673307
2 55635 DEPDC1 17.3_VEH_0.5_24 4 VEH 0.5 24 4.912021
3 55635 DEPDC1 84.1_CX.5461_0.5_24 5 CX.5461 0.5 24 2.736395
4 55635 DEPDC1 84.1_VEH_0.5_24 5 VEH 0.5 24 4.149703
5 55635 DEPDC1 90.1_CX.5461_0.5_24 6 CX.5461 0.5 24 2.430108
6 55635 DEPDC1 90.1_VEH_0.5_24 6 VEH 0.5 24 4.224172📌Generate Boxplot
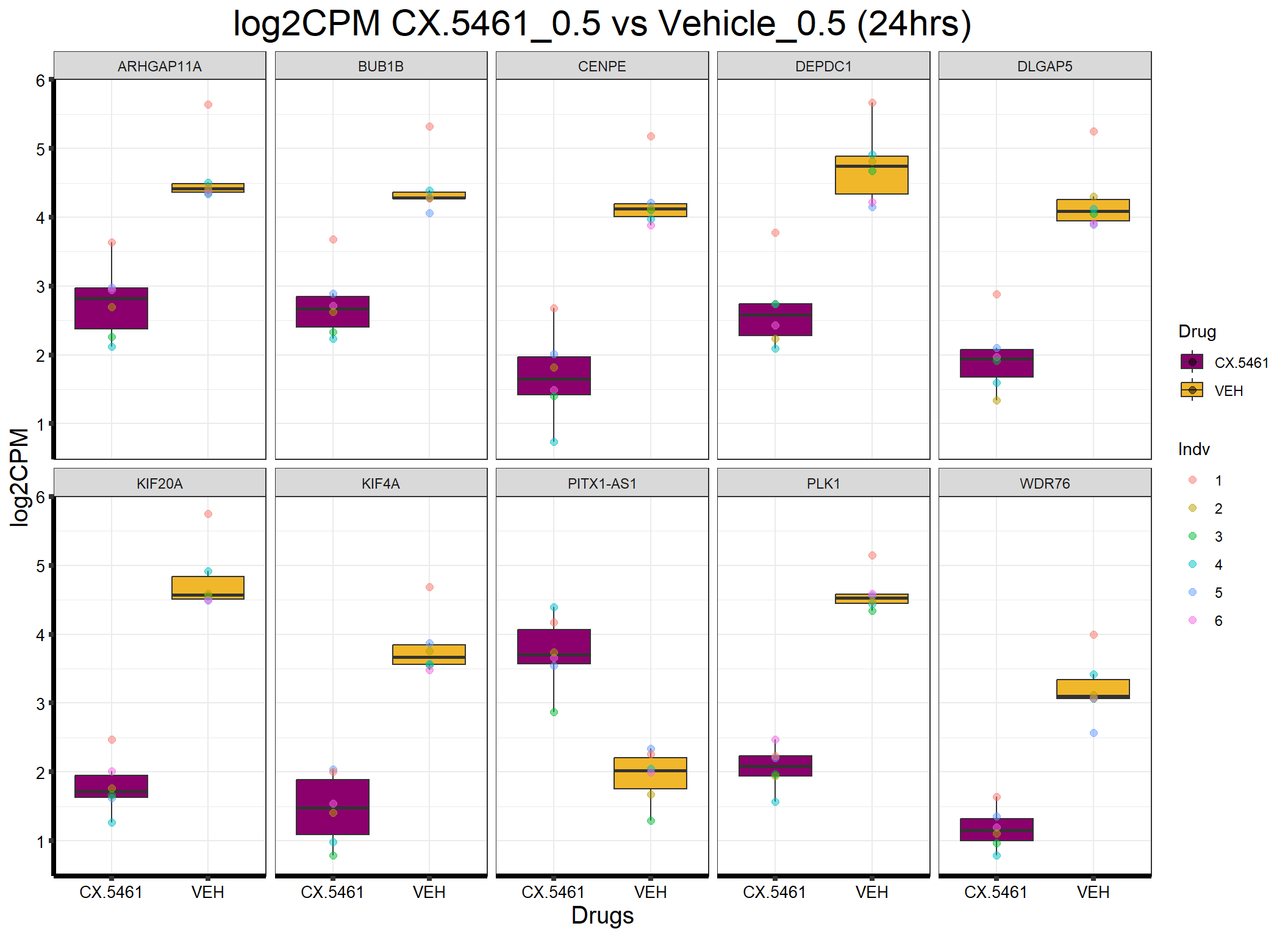
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM CX.5461_0.5 vs Vehicle_0.5 (24hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 CX_0.5_48 vs VEH_0.5_48
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_CX_0.5_48.csv"
output_boxplot <- "data/Boxplot_CX_0.5_48.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|CX.5461_0.5_48|VEH_0.5_48", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("CX.5461" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 4751 -6.214248 1.300150 -16.68265 5.33e-30 7.60e-26 57.15221
2 55635 -4.249490 3.616797 -15.29901 2.20e-27 1.04e-23 51.73471
3 1062 -3.886212 2.975691 -15.36246 1.66e-27 1.04e-23 51.99052
4 9787 -4.888829 3.045935 -15.12843 4.69e-27 1.67e-23 50.81204
5 5347 -3.890052 3.460559 -14.92237 1.18e-26 3.37e-23 50.09400
6 701 -2.665736 3.456542 -14.53166 6.87e-26 1.40e-22 48.42019📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("CX.5461", Sample), "CX.5461", "VEH"),
Conc = "0.5",
Timepoint = "48"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 55635 DEPDC1 87.1_CX.5461_0.5_48 3 CX.5461 0.5 48 0.5823875
2 55635 DEPDC1 87.1_VEH_0.5_48 3 VEH 0.5 48 5.1235645
3 55635 DEPDC1 17.3_CX.5461_0.5_48 4 CX.5461 0.5 48 0.7666322
4 55635 DEPDC1 17.3_VEH_0.5_48 4 VEH 0.5 48 4.9751550
5 55635 DEPDC1 84.1_CX.5461_0.5_48 5 CX.5461 0.5 48 0.7510937
6 55635 DEPDC1 84.1_VEH_0.5_48 5 VEH 0.5 48 5.0865557📌Generate Boxplot
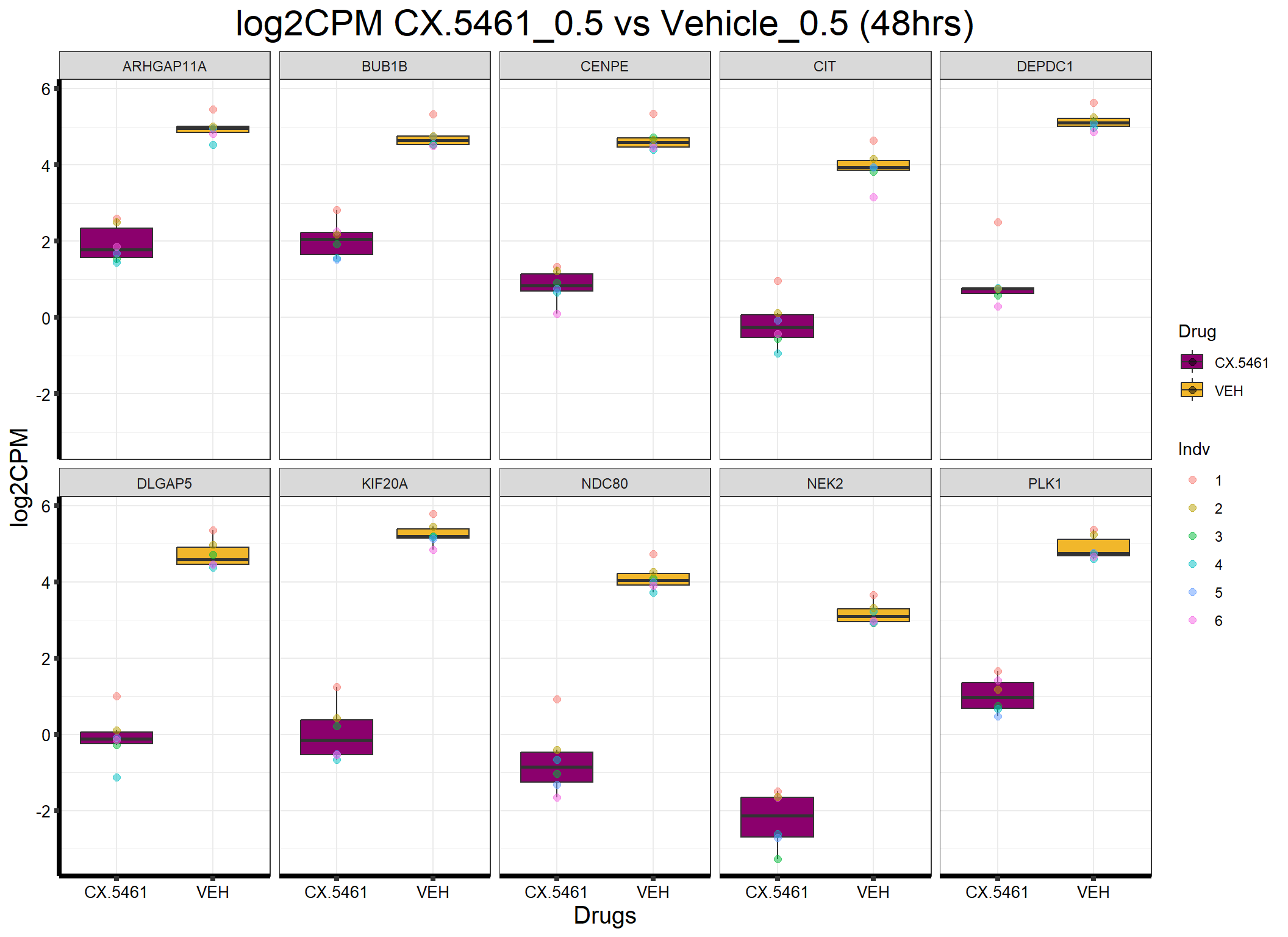
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM CX.5461_0.5 vs Vehicle_0.5 (48hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 DOX_0.1_3 vs VEH_0.1_3
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_DOX_0.1_3.csv"
output_boxplot <- "data/Boxplot_DOX_0.1_3.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|DOX_0.1_3|VEH_0.1_3", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("DOX" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 4899 -0.6551523 3.538531 -5.605223 2.03e-07 0.002891627 -0.69950274
2 51278 0.6502711 5.807772 4.928006 3.51e-06 0.025032033 0.04876888📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("DOX", Sample), "DOX", "VEH"),
Conc = "0.1",
Timepoint = "3"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 51278 IER5 84.1_DOX_0.1_3 5 DOX 0.1 3 5.856020
2 51278 IER5 84.1_VEH_0.1_3 5 VEH 0.1 3 6.105139
3 51278 IER5 17.3_DOX_0.1_3 4 DOX 0.1 3 6.250617
4 51278 IER5 90.1_DOX_0.1_3 6 DOX 0.1 3 6.030993
5 51278 IER5 90.1_VEH_0.1_3 6 VEH 0.1 3 5.305258
6 51278 IER5 17.3_VEH_0.1_3 4 VEH 0.1 3 5.448695📌Generate Boxplot
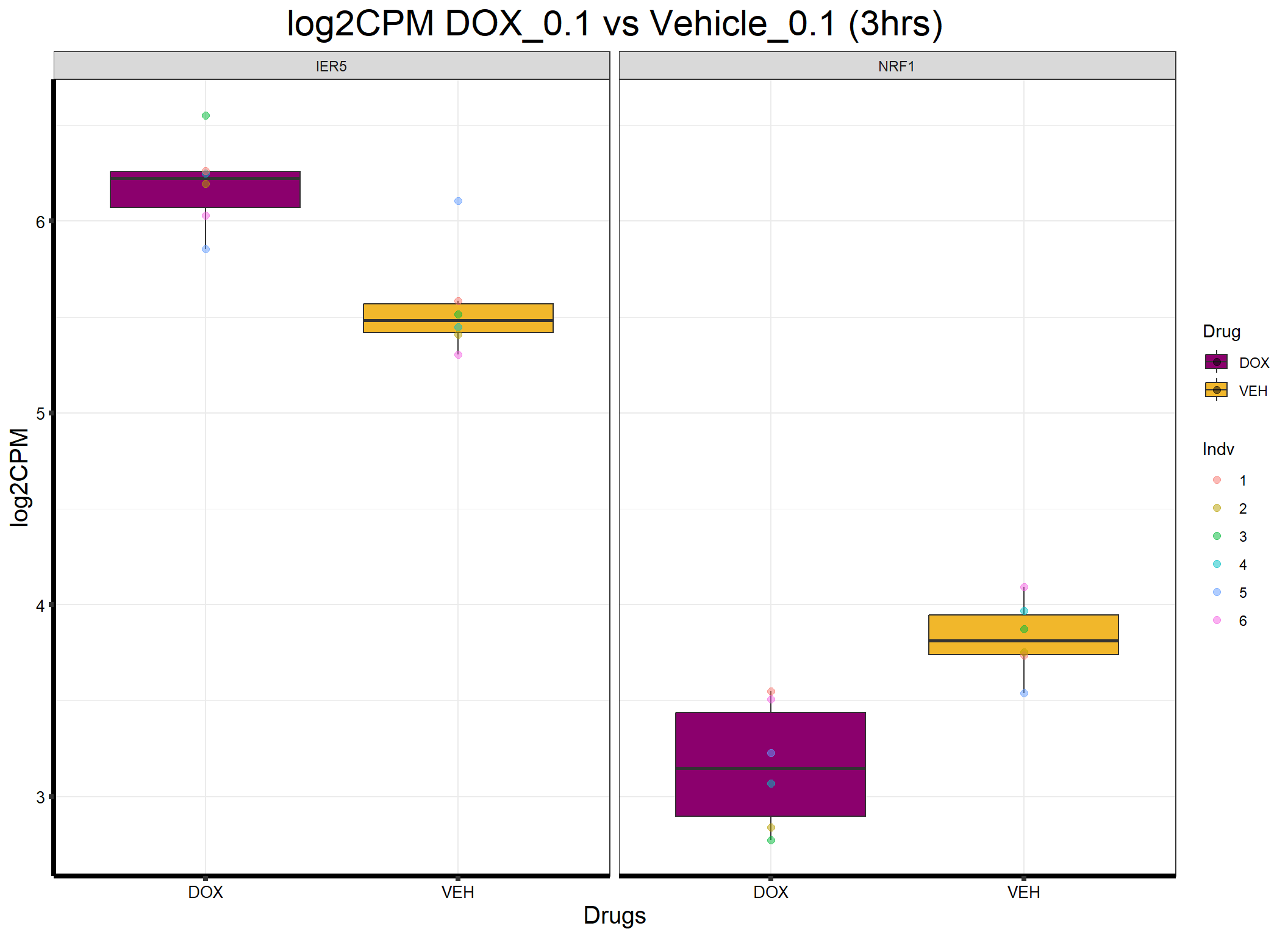
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM DOX_0.1 vs Vehicle_0.1 (3hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 DOX_0.1_24 vs VEH_0.1_24
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_DOX_0.1_24.csv"
output_boxplot <- "data/Boxplot_DOX_0.1_24.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|DOX_0.1_24|VEH_0.1_24", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("DOX" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 701 -2.685154 3.456542 -14.59080 5.25e-26 4.54e-22 48.56697
2 5347 -3.571951 3.460559 -14.54877 6.35e-26 4.54e-22 48.28765
3 1062 -3.445122 2.975691 -14.13731 4.14e-25 1.97e-21 46.39529
4 55635 -3.271838 3.616797 -13.47297 8.91e-24 3.18e-20 43.52573
5 9787 -3.592795 3.045935 -13.14501 4.13e-23 1.18e-19 41.91228
6 9824 -2.588518 3.672008 -12.80816 2.02e-22 4.12e-19 40.50264📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("DOX", Sample), "DOX", "VEH"),
Conc = "0.1",
Timepoint = "24"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 55635 DEPDC1 87.1_VEH_0.1_24 3 VEH 0.1 24 4.942031
2 55635 DEPDC1 17.3_VEH_0.1_24 4 VEH 0.1 24 4.691377
3 55635 DEPDC1 84.1_DOX_0.1_24 5 DOX 0.1 24 1.559433
4 55635 DEPDC1 84.1_VEH_0.1_24 5 VEH 0.1 24 4.869754
5 55635 DEPDC1 90.1_DOX_0.1_24 6 DOX 0.1 24 1.077164
6 55635 DEPDC1 90.1_VEH_0.1_24 6 VEH 0.1 24 4.176633📌Generate Boxplot
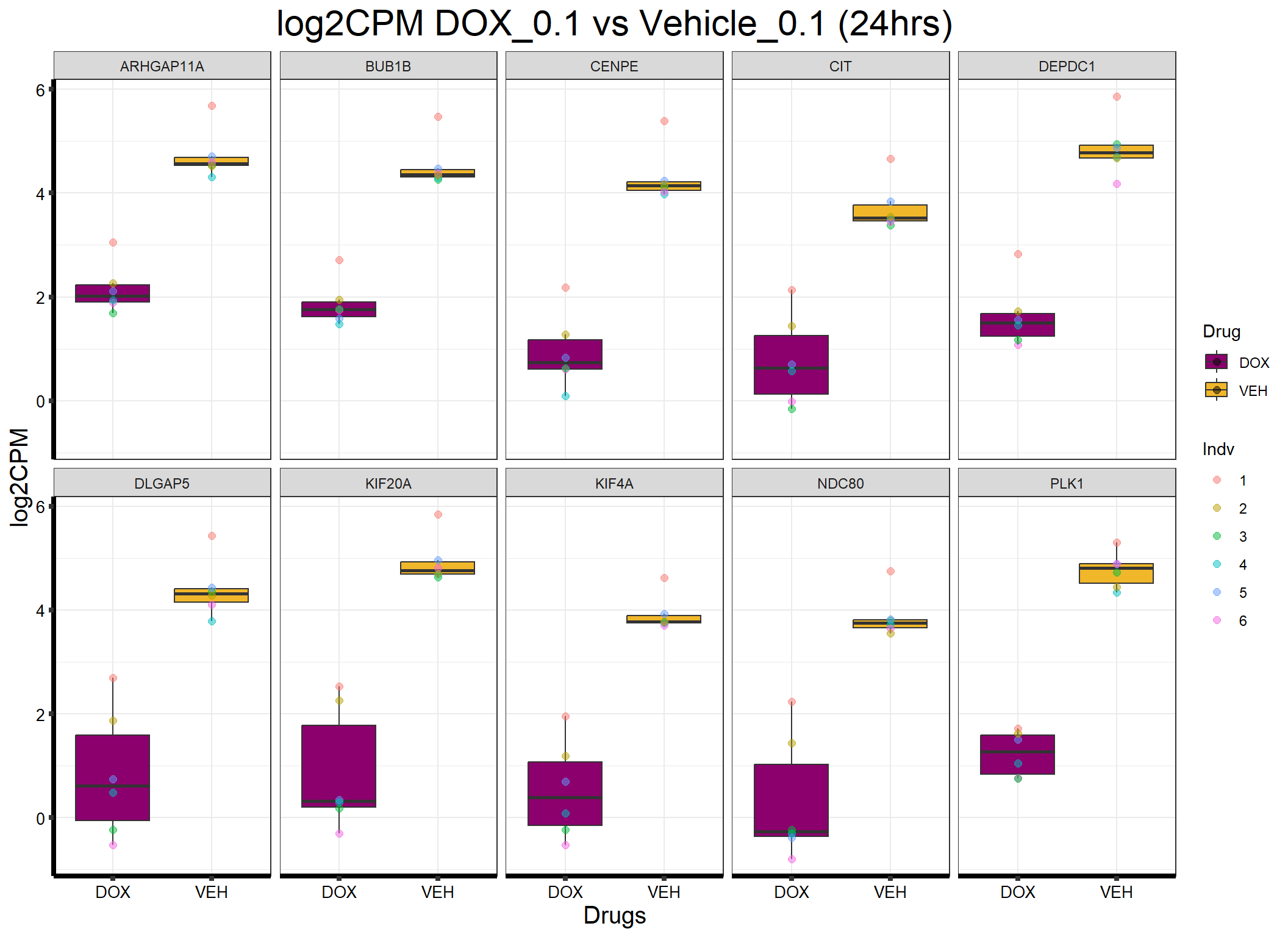
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM DOX_0.1 vs Vehicle_0.1 (24hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 DOX_0.1_48 vs VEH_0.1_48
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_DOX_0.1_48.csv"
output_boxplot <- "data/Boxplot_DOX_0.1_48.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|DOX_0.1_48|VEH_0.1_48", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("DOX" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 4751 -6.816249 1.3001500 -18.39628 4.30e-33 3.07e-29 64.48879
2 57405 -5.671870 0.7274157 -18.40627 4.13e-33 3.07e-29 64.48778
3 10403 -6.252449 2.3623660 -18.27841 6.94e-33 3.30e-29 64.04498
4 9787 -6.197811 3.0459352 -17.69659 7.53e-32 2.69e-28 61.73461
5 83540 -5.473128 1.7658990 -17.26718 4.50e-31 1.10e-27 59.97652
6 55872 -6.962669 2.8078976 -17.26122 4.61e-31 1.10e-27 59.96853📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("DOX", Sample), "DOX", "VEH"),
Conc = "0.1",
Timepoint = "48"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 55635 DEPDC1 87.1_DOX_0.1_48 3 DOX 0.1 48 0.1715850
2 55635 DEPDC1 87.1_VEH_0.1_48 3 VEH 0.1 48 5.5921101
3 55635 DEPDC1 17.3_DOX_0.1_48 4 DOX 0.1 48 -0.2683688
4 55635 DEPDC1 17.3_VEH_0.1_48 4 VEH 0.1 48 5.0422478
5 55635 DEPDC1 84.1_DOX_0.1_48 5 DOX 0.1 48 0.3643567
6 55635 DEPDC1 84.1_VEH_0.1_48 5 VEH 0.1 48 4.9304649📌Generate Boxplot
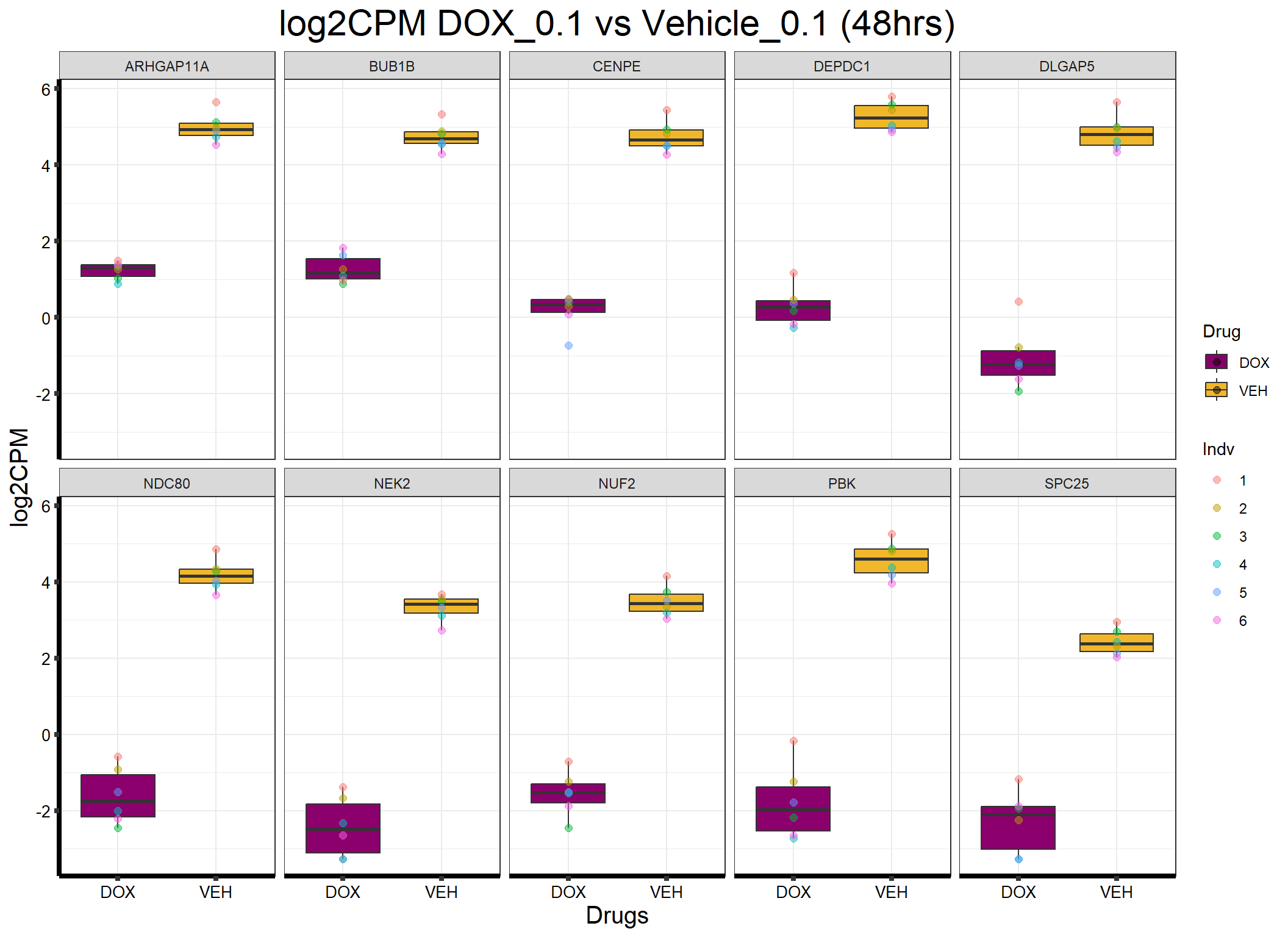
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM DOX_0.1 vs Vehicle_0.1 (48hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 DOX_0.5_3 vs VEH_0.5_3
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_DOX_0.5_3.csv"
output_boxplot <- "data/Boxplot_DOX_0.5_3.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|DOX_0.5_3|VEH_0.5_3", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("DOX" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 10221 -1.8256324 6.540834 -8.201574 1.12e-12 1.60e-08 18.45455
2 114821 -1.3410345 2.661316 -7.855281 6.03e-12 4.30e-08 16.22434
3 84872 -1.0552959 2.733403 -7.670032 1.47e-11 5.26e-08 15.36902
4 57547 -1.4472714 1.953625 -7.682170 1.39e-11 5.26e-08 15.12367
5 4800 -0.7830463 4.914152 -7.351552 6.78e-11 1.94e-07 14.48395
6 4091 -0.8956369 6.397947 -6.989048 3.78e-10 7.49e-07 12.87543📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("DOX", Sample), "DOX", "VEH"),
Conc = "0.5",
Timepoint = "3"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 84872 ZC3H10 84.1_DOX_0.5_3 5 DOX 0.5 3 2.690727
2 84872 ZC3H10 84.1_VEH_0.5_3 5 VEH 0.5 3 2.511475
3 84872 ZC3H10 17.3_DOX_0.5_3 4 DOX 0.5 3 1.056370
4 84872 ZC3H10 90.1_DOX_0.5_3 6 DOX 0.5 3 2.213516
5 84872 ZC3H10 90.1_VEH_0.5_3 6 VEH 0.5 3 3.019635
6 84872 ZC3H10 75.1_DOX_0.5_3 1 DOX 0.5 3 1.862865📌Generate Boxplot
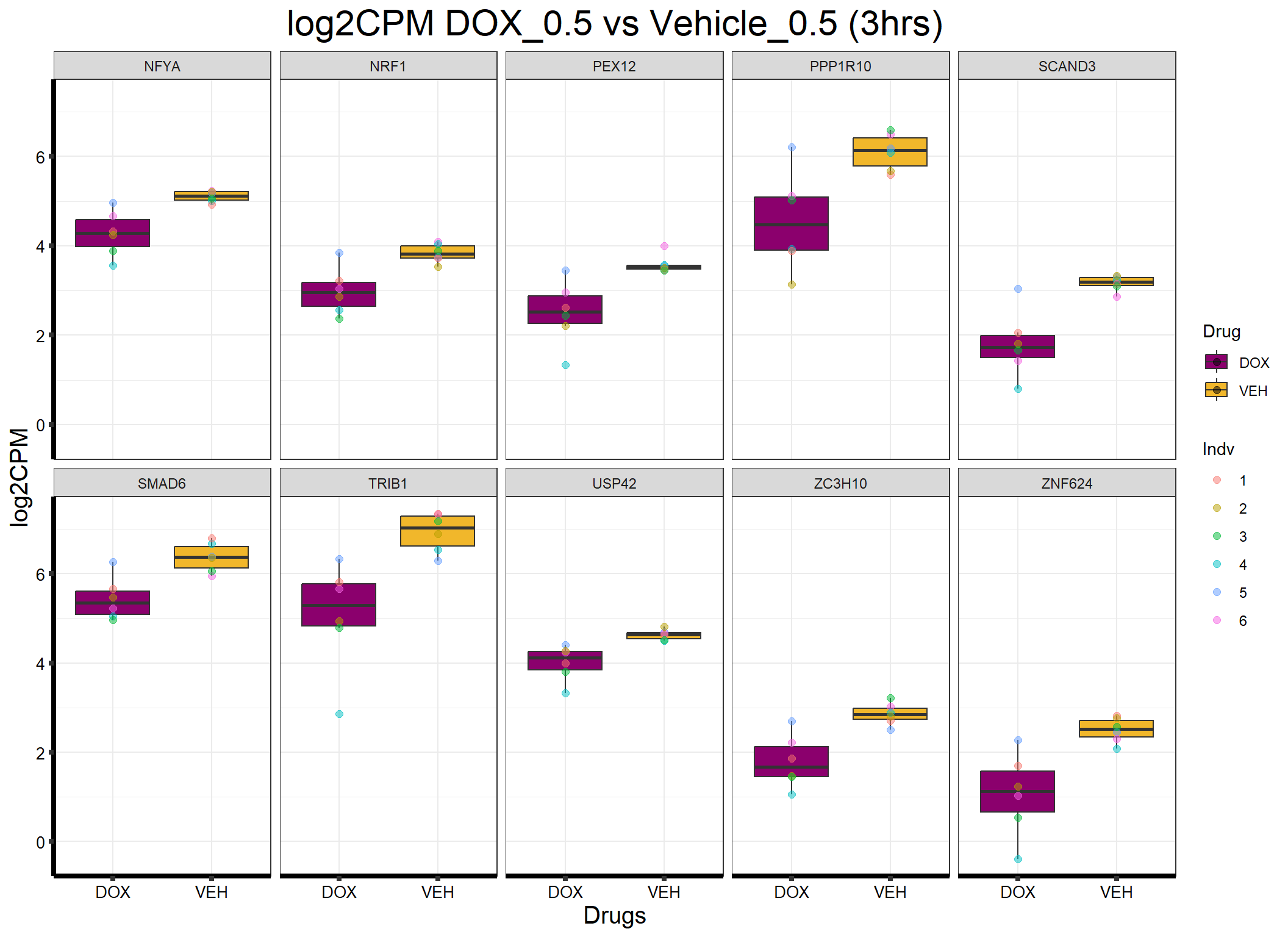
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM DOX_0.5 vs Vehicle_0.5 (3hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 DOX_0.5_24 vs VEH_0.5_24
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_DOX_0.5_24.csv"
output_boxplot <- "data/Boxplot_DOX_0.5_24.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|DOX_0.5_24|VEH_0.5_24", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("DOX" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 3065 -1.393169 5.630830 -15.69290 3.86e-28 5.51e-24 53.54984
2 861 3.015106 3.533037 15.31790 2.02e-27 1.44e-23 51.85161
3 7091 1.959276 4.291890 14.38780 1.32e-25 6.28e-22 47.77567
4 9695 2.223504 5.281472 13.87772 1.37e-24 4.87e-21 45.45873
5 104472848 4.137451 1.699347 13.74711 2.50e-24 7.13e-21 44.64516
6 6382 3.987394 5.024872 13.63845 4.13e-24 9.83e-21 44.37005📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("DOX", Sample), "DOX", "VEH"),
Conc = "0.5",
Timepoint = "24"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 3065 HDAC1 17.3_DOX_0.5_24 4 DOX 0.5 24 4.746736
2 3065 HDAC1 87.1_DOX_0.5_24 3 DOX 0.5 24 4.379176
3 3065 HDAC1 87.1_VEH_0.5_24 3 VEH 0.5 24 5.887956
4 3065 HDAC1 17.3_VEH_0.5_24 4 VEH 0.5 24 5.971019
5 3065 HDAC1 84.1_DOX_0.5_24 5 DOX 0.5 24 4.920790
6 3065 HDAC1 84.1_VEH_0.5_24 5 VEH 0.5 24 5.813260📌Generate Boxplot
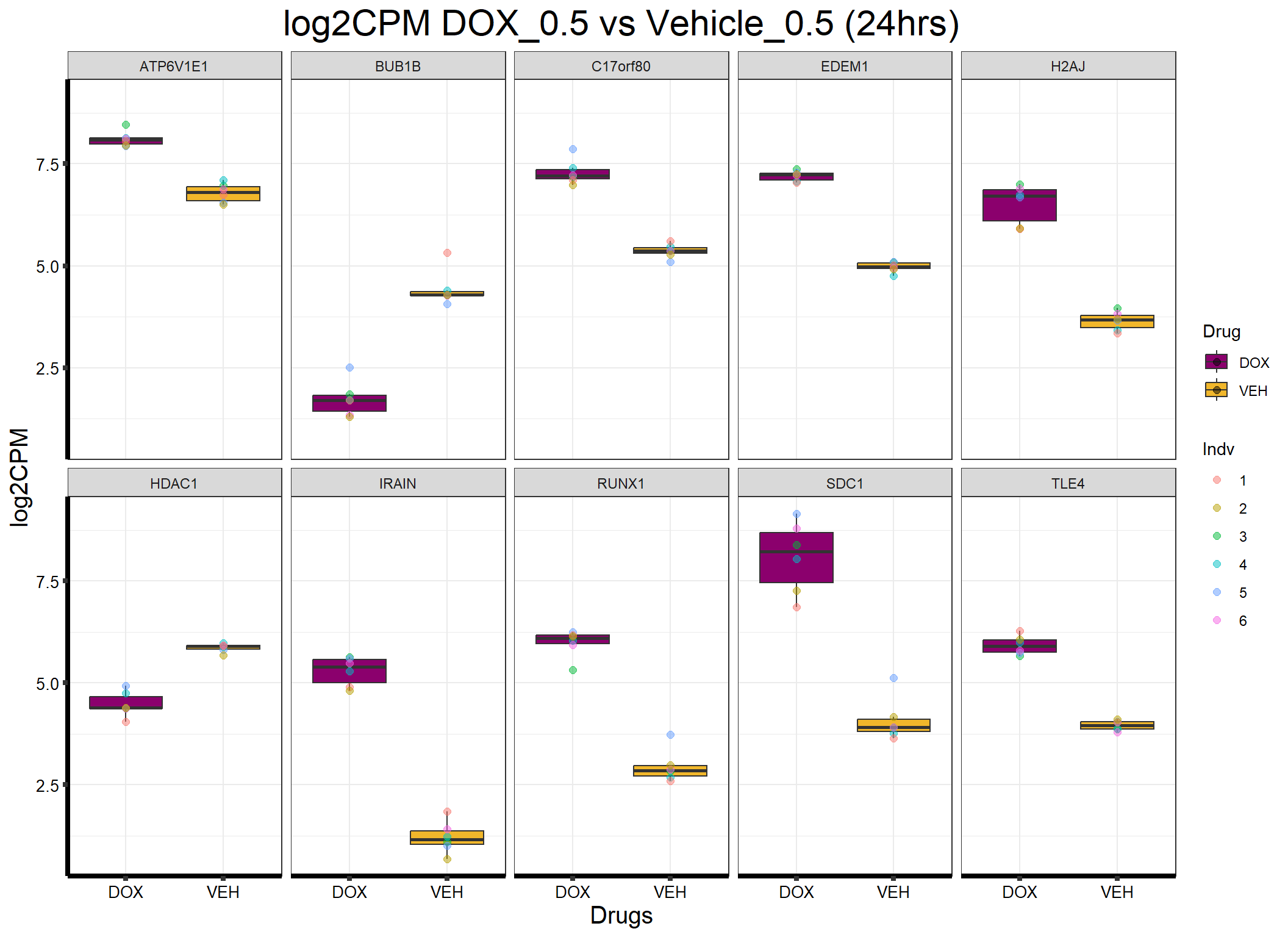
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM DOX_0.5 vs Vehicle_0.5 (24hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
📌 DOX_0.5_48 vs VEH_0.5_48
📌 Define File Paths
# Define file paths
input_file <- "data/Feature_count_Matrix_Log2CPM_filtered.csv"
toptable_file <- "data/DEGs/Toptable_DOX_0.5_48.csv"
output_boxplot <- "data/Boxplot_DOX_0.5_48.csv"
output_log2CPM <- "data/log2CPM_filtered.csv"📌 Read and Clean Dataset
# Read Log2CPM Matrix
boxplot1 <- read.csv(input_file) %>%
as.data.frame()
colnames(boxplot1) <- trimws(gsub("^X", "", colnames(boxplot1))) # Clean column names
# Select relevant columns
matching_columns <- grep("ENTREZID|SYMBOL|DOX_0.5_48|VEH_0.5_48", colnames(boxplot1), value = TRUE)
if (!"SYMBOL" %in% colnames(boxplot1)) stop("ERROR: 'SYMBOL' column is missing in boxplot1! Check dataset.")
new_boxplot1 <- boxplot1[, matching_columns]
# Save cleaned datasets
write.csv(boxplot1, output_log2CPM, row.names = FALSE)
write.csv(new_boxplot1, output_boxplot, row.names = FALSE)
# Display first few rows
head(new_boxplot1)📌 Define Color Palettes
drug_palc <- c("DOX" = "#8B006D", "VEH" = "#F1B72B")📌 Read and Process Toptable Data
toptable <- read.csv(toptable_file)
colnames(toptable)[1] <- "Entrezid"
filtered_toptable <- toptable %>%
filter(adj.P.Val < 0.05) %>%
arrange(adj.P.Val) %>%
slice_head(n = 10)
# Display first few rows of the filtered Toptable
head(filtered_toptable) Entrezid logFC AveExpr t P.Value adj.P.Val B
1 79837 1.790008 4.956669 18.06416 1.66e-32 1.19e-28 63.51059
2 701 -4.137116 3.456542 -18.17980 1.04e-32 1.19e-28 63.01482
3 10376 -2.133871 8.392149 -16.54422 9.62e-30 2.75e-26 57.22623
4 2013 -3.053995 8.888062 -16.57290 8.51e-30 2.75e-26 57.34618
5 7091 2.243767 4.291890 16.54672 9.52e-30 2.75e-26 57.20487
6 1917 -2.842549 9.408686 -16.38606 1.90e-29 4.51e-26 56.55109📌 Filter Boxplot Data for Selected Genes
filtered_boxplot <- new_boxplot1 %>%
filter(ENTREZID %in% filtered_toptable$Entrezid)
if (!"SYMBOL" %in% colnames(filtered_boxplot)) stop("ERROR: 'SYMBOL' column is missing in filtered_boxplot!")
# Pivot data to long format
long_boxplot <- filtered_boxplot %>%
pivot_longer(cols = -c(ENTREZID, SYMBOL), names_to = "Sample", values_to = "log2CPM") %>%
as.data.frame()
if (!"SYMBOL" %in% colnames(long_boxplot)) stop("ERROR: 'SYMBOL' column is missing after pivoting!")
long_boxplot$SYMBOL <- as.character(long_boxplot$SYMBOL)📌 Define Individual Mapping
indv_mapping <- c("75.1" = 1, "78.1" = 2, "87.1" = 3, "17.3" = 4, "84.1" = 5, "90.1" = 6)
# Debug: Print column names before mutate
print("Columns in long_boxplot before mutate():")[1] "Columns in long_boxplot before mutate():"print(colnames(long_boxplot))[1] "ENTREZID" "SYMBOL" "Sample" "log2CPM" 📌Process Data with Metadata
formatted_data <- long_boxplot %>%
mutate(
Indv_id = sub("_.*", "", Sample), # Extract individual ID
Indv = ifelse(Indv_id %in% names(indv_mapping), as.character(indv_mapping[Indv_id]), NA),
Drug = ifelse(grepl("DOX", Sample), "DOX", "VEH"),
Conc = "0.5",
Timepoint = "48"
) %>%
dplyr::select(any_of(c("ENTREZID", "SYMBOL", "Sample", "Indv", "Drug", "Conc", "Timepoint", "log2CPM"))) %>%
as.data.frame()
# Rename SYMBOL → Gene
colnames(formatted_data) <- trimws(colnames(formatted_data))
if ("SYMBOL" %in% colnames(formatted_data)) {
colnames(formatted_data)[colnames(formatted_data) == "SYMBOL"] <- "Gene"
} else {
stop("ERROR: 'SYMBOL' column is missing before renaming.")
}
if (!"Gene" %in% colnames(formatted_data)) stop("ERROR: 'Gene' column is missing after renaming.")
formatted_data$Indv <- as.character(formatted_data$Indv)
# Display first few rows
head(formatted_data) ENTREZID Gene Sample Indv Drug Conc Timepoint log2CPM
1 10376 TUBA1B 87.1_DOX_0.5_48 3 DOX 0.5 48 6.296386
2 10376 TUBA1B 87.1_VEH_0.5_48 3 VEH 0.5 48 8.699481
3 10376 TUBA1B 17.3_DOX_0.5_48 4 DOX 0.5 48 6.996916
4 10376 TUBA1B 17.3_VEH_0.5_48 4 VEH 0.5 48 8.780009
5 10376 TUBA1B 84.1_DOX_0.5_48 5 DOX 0.5 48 6.906286
6 10376 TUBA1B 84.1_VEH_0.5_48 5 VEH 0.5 48 8.714215📌Generate Boxplot
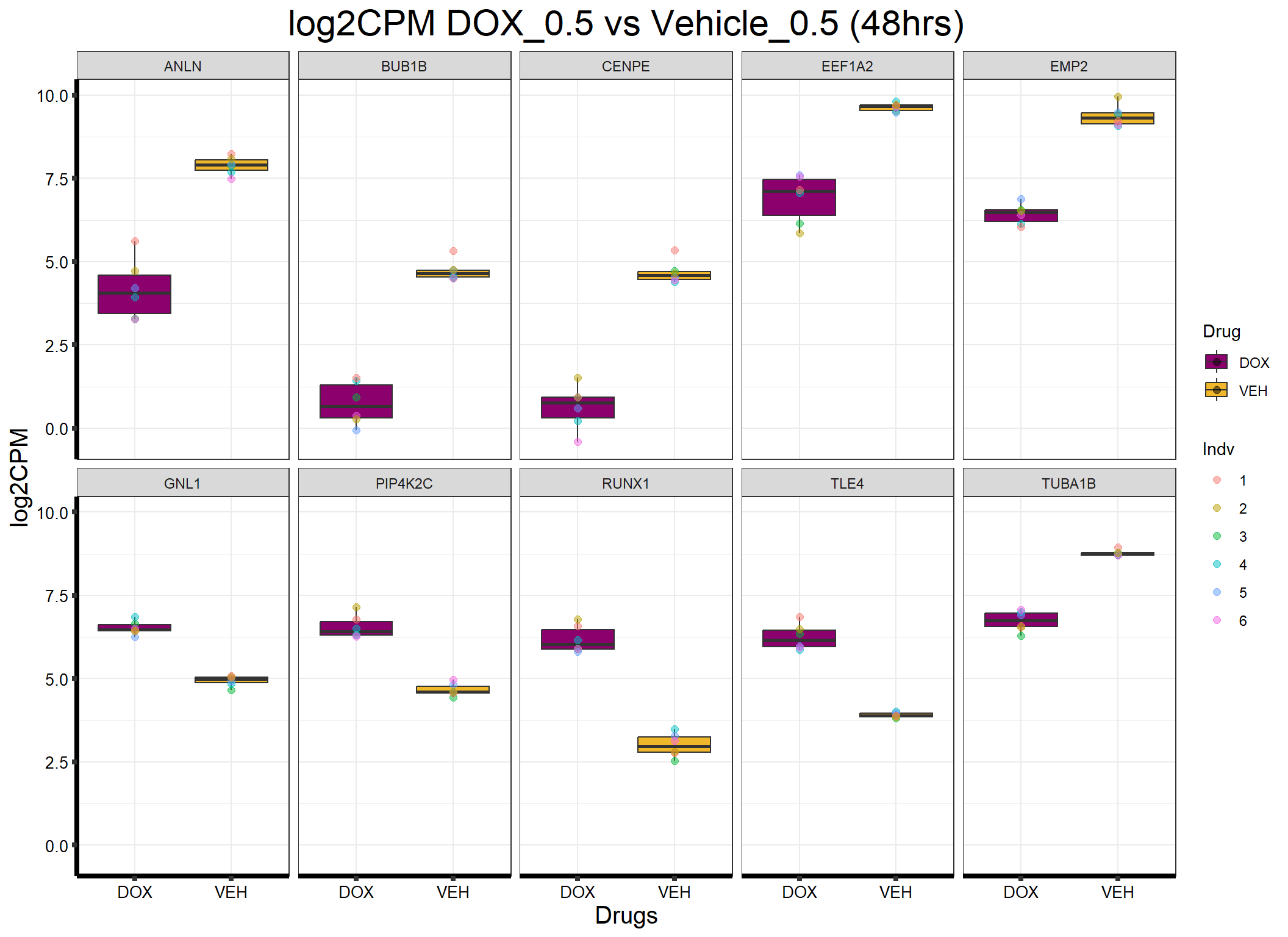
ggplot(formatted_data, aes(x = Drug, y = log2CPM, fill = Drug)) +
geom_boxplot(outlier.shape = NA) +
scale_fill_manual(values = drug_palc) +
facet_wrap(~ Gene, ncol = 5) +
geom_point(aes(color = Indv), size = 2, alpha = 0.5, position = position_jitter(width = -1, height = 0)) +
ggtitle("log2CPM DOX_0.5 vs Vehicle_0.5 (48hrs)") +
labs(x = "Drugs", y = "log2CPM") +
theme_bw() +
theme(
plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size = 10, color = "black"),
axis.text.x = element_text(size = 10, color = "black"),
strip.text.y = element_text(color = "white")
)
sessionInfo()R version 4.3.0 (2023-04-21 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 11 x64 (build 22631)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidyr_1.3.1 ggsignif_0.6.4 ggplot2_3.5.1 dplyr_1.1.4
[5] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] gtable_0.3.6 jsonlite_1.8.9 compiler_4.3.0 promises_1.3.0
[5] tidyselect_1.2.1 Rcpp_1.0.12 stringr_1.5.1 git2r_0.35.0
[9] callr_3.7.6 later_1.3.2 jquerylib_0.1.4 scales_1.3.0
[13] yaml_2.3.10 fastmap_1.1.1 R6_2.5.1 labeling_0.4.3
[17] generics_0.1.3 knitr_1.49 tibble_3.2.1 munsell_0.5.1
[21] rprojroot_2.0.4 bslib_0.8.0 pillar_1.10.1 rlang_1.1.3
[25] cachem_1.0.8 stringi_1.8.3 httpuv_1.6.15 xfun_0.50
[29] getPass_0.2-4 fs_1.6.3 sass_0.4.9 cli_3.6.1
[33] withr_3.0.2 magrittr_2.0.3 ps_1.8.1 grid_4.3.0
[37] digest_0.6.34 processx_3.8.5 rstudioapi_0.17.1 lifecycle_1.0.4
[41] vctrs_0.6.5 evaluate_1.0.3 glue_1.7.0 farver_2.1.2
[45] whisker_0.4.1 colorspace_2.1-0 purrr_1.0.2 rmarkdown_2.29
[49] httr_1.4.7 tools_4.3.0 pkgconfig_2.0.3 htmltools_0.5.8.1